生物資料庫簡介
蛋白質序列結構的分析與預測
口述:呂平江老師
整理:曾瓘鈞、林泰宏
廖智凱、李岳錡
2.PROTEIN IDENTITY BASED ON COMPOSTION
3.AACompldent and AACompSim(ExPASy)
5.MOWSE
6.PHYSICAL PROPERTIES BASED ON SEQUENCE
這本書之前的章節關於資料庫得到知識的討論,可以運用不同的資料庫得到大量可用的序列訊息,當我們準備看核甘酸序列及所有的蛋白質序列時,無論是直接決定,或是經由核甘酸序列中open reading frame的轉譯,這些包含決定其結構及功能的內在訊息,不幸的,實驗針對這些訊息不能用未加工的訊息資料來產生,一些判定的技術,像是circular dichroism spectroscopy、optical rototary dispersion、 X-ray晶體繞射(X-ray crystallography)及核磁共振(NMR),對於結構的特性是非常強而有力,但這些費時的技術實行,需要高度熟練和技術性上高要求的操作,在蛋白質序列和結構資料庫的大小上比較中,SWISS-PORT中有87143個蛋白質(Release 39.0),但只有12624的蛋白質結構在PDB中出現(July, 2000),試圖關掉環繞在預測結構跟功能的方法中的gap center,然後這些方式可以在生化資料缺乏時,提供一個看的見蛋白質特性的方法。
此章節焦點集中在計算的技術,可以提供學上的發現基於本身蛋白質序列或其本身蛋白質家族的比較,不像核甘酸序列,是由4個化學上相似的base所組成,蛋白質中找到20個胺基酸,提供了結構及功能非常大的變異,這些殘基具有不同的化學構造,因為胺基酸是鹼或是酸、是親水性或是厭水性、還是直鍊或是具有分支鏈、或是芳香族,所以每一個殘基皆可影響蛋白質全部物理特性,因此,在蛋白質domain上,每一個殘基具有某一傾向去形成不同型的結構,這些特性,基於一個生化中心的教條:序列詳述構造。
不管用何種預測性的技術,它的結果都只是預測,不同的方法,用不同的規則系統,或許是、或許不是我們所預測的結果,重要的是,如何去操作一個特有的預測方法,而不是系統規則上的黑盒子:一個方法或許是適用一個特有的事件,但不能完全適用於另一個事件,即使如此,適當的利用這些技術和初期的生化資料,可以對於蛋白質結構及功能上提供有價值的鑑識。
2.PROTEIN IDENTITY BASED ON COMPOSTION
20個胺基酸的物理及化學特性完全的了解,基於這些特性,許多有用的計算工具,已經發展用來預測未知蛋白質的辨別,在the Swiss Institute of Bioinformatics中,很多工具經由ExPASy server獲得。
ExPASy的焦點是雙倍功能,一個經由2-D電泳分離的未知蛋白質之協助分析,和預測已知蛋白質的未知特性,這些利用SWISS-PORT的註解來作它們的預測,雖然計算結果例如在電泳分析上是有用的,但是在很多的實驗範圍上是有價值的,例如一些色層分析和沉澱分析的研究,在這這段落及接下來的段落,在這個ExPASy suite中是可被辨識的,但是接下來的討論也包含很多有用的程式,包括許多有用的程式internet資源和這些工具的關聯,會在這個章節陸續列出來討論。
3.AACompldent and AACompSim(ExPASy)
與其利用胺基酸序列去搜尋SWISS-PORT,還不如AACompldent用未知蛋白質胺基酸的組成去辨識是同似組成的已知蛋白質,當輸入時程式,需要合適胺基酸組成、等電點(pI)、和蛋白質的分子量(如果已知)、適當的分類taxonomic class和任何特殊的關鍵字,除此之外,使用者必須選擇6個胺基酸中的一個(constellations),這個可以影響分析如何執行,例如某一群(constellations)結合殘基像似Asp╱Asn(D╱N)、Gln╱Glu(Q╱E)變成為Asx(B)、Glx(Z),或某些殘基從分析中完全被除去,為了在資料庫每一個序列,規則系統基於不同的序列和查詢的組成之間,計算出分數,這個的結果可以用e-mail答覆,其中包含三個等級的列表:
![]() 一個列表基於從記載從taxonomic
class來的所有蛋白質,不用考慮pI或蛋白質的分子量。
一個列表基於從記載從taxonomic
class來的所有蛋白質,不用考慮pI或蛋白質的分子量。
![]() 一個列表基於所有的蛋白質,不管taxonomic
class,不用考慮pI或蛋白質的分子量。
一個列表基於所有的蛋白質,不管taxonomic
class,不用考慮pI或蛋白質的分子量。
![]() 一個列表基於記載的taxonomic
class但是要考慮pI和蛋白質的分子量。
一個列表基於記載的taxonomic
class但是要考慮pI和蛋白質的分子量。
因為計算的分數是不同的測定,分數為零暗只在查詢組成按序列記載之間的有正確一致性。
AACompSim,它是AACompldent的變形,執行分析的類似型態,但是,與其利用實驗上來的胺基酸組成作基本的組成搜尋,還不如用SWISS-PORT protein的序列,在不同分數的計算用於Compute pI╱MW之前,理論上的pI和分子量先被計算,它可以提供證據指出跨越種界線的胺基酸是可以很好的被保留下來,而且藉由考慮胺基酸的組成,研究員可以偵測蛋白質間的微弱相似性,(這些蛋白質序列falls below 25﹪),因此,除了執行典型的資料庫搜尋之外,組成的考慮可以提供暫蛋白質之間,提供額外的鑑識。
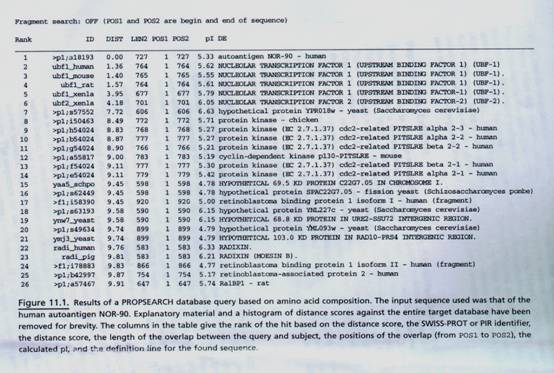
順著像AACompSim的同路徑,PROPSEARCH利用蛋白質的胺基酸組成,去偵測蛋白質之間微弱的關係,而且作者已經說明這個技術,可以簡單地被用於去辨識相同protein family的成員,然而,這個技術比具有144個物理特性的AACompSim來的費力,而被用於分析其中包含分子量案大量殘基的內含物厭水值和平均電荷,這個物理特性的蒐集稱為查詢載體(query vector),而且,在目標資料庫中,先計算每一個相同型態載體可以被比較(SWISS-PORT和PIR)這個載體的資料庫的計算有利於促進查詢的過程時間,輸入PROPSEARCH Web server可以查詢序列,程式輸豬的例子例如圖11.1這裡human autoantigen NOR-90的序列,被用於作輸入的查詢,這個結果可以藉由distance score被分類,然後這個score代表經由PROPSEARCH找到查詢序列和新序列屬於相同family可能性,因此,在多數的事件中暗示共同的功能,distance score為10或10以下,指出大於87﹪的機會在兩個蛋白質之間是有相似性的,而這個分數低於於8.7,可增加可靠性達94﹪,而分數低於7.5則,可增加可靠性達99.6﹪,這個結果的檢查顯示NOR-90它和很多核仁轉錄因子、protein kinases、a retinoblastoma-binding protein、the actin-binding protein radixin,和RalBP1、假定的GTPase target有相似性,一但這些蛋白質的功能不相似時,沒有任何的hit需要被預期的;然而,這些大多數為DNA-binding protein,這些蛋白質可以打開相似domain的可能性而被運用兩者選一的功能上之狀況,至少,BLASTP search對於證明結果和確認critical residues是不可或缺的。
5.MOWSE
The Molecular Weight Search(MOWSE)的運算法則,是利用mass spectrometric(MS)techniques所得到的訊息,完整的蛋白質分子量和一些因用特殊的protease得到的相同蛋白質的分解物,都可以一起使用,給予數個實驗測定的結果,一個未知的蛋白質可以很清晰的被確認,一但未知蛋白質沒有完整或部分被定序,這個方法大致上可以縮減實驗的時間。
和經由試劑所產生的the resultant masses以及peptide的組成一樣,The MOWSE Web front end需要一開始序列的分子量和化學試劑的使用,a tolerance value可能被記載,指出在決定the determined fragment massese的正確性中的錯誤許可,計算方式基於the OWL nonredudant protein sequence database中的訊息,得分基於如何在分子量給予範圍內的蛋白質中,片段分子量的存在,而輸出是回復分類列表頂端30個分數,用OWL entry the name、相配的peptide序列和其他統計上的知識,simulation studies用5個或較少的輸入peptide重量可以產生99﹪的正確率。
6.PHYSICAL PROPERTIES BASED ON SEQUENCE
Compute pI╱MW and ProtParam(ExPASy)
Compute pI╱MW這個工具可以計算輸入序列的等電點和分子量,pI的決定基於pK值,描述從中性到酸性pH值的變性環境中,對於蛋白質遷徙的研究,因為這個原因,作者警告pI對於鹼性蛋白質的測定是不正確的,分子量藉由在序列中,每一個胺基酸的平均isotopic mass添加,再加上一個水分子,藉由這些東西被計算出來,藉由FASTA格式化中的使用者、或a SWISS-PORT identifier、或加入數目,序列可以被供應而被記載,假如序列是被供給的,工具可以自動計算晚整的序列長度的pI值和分子量,如果SWISS-PORT identifier是被給予的,進去的定義和生物路徑是被顯示出來的,而使用者可以詳述胺基酸的範圍,已至於計算在片段而不是在完整的蛋白質中被做出來,ProtParam它是更進一步的過程,根據輸入的序列,ProtParam計算分子量、等電點、全部的胺基酸組成、理論上的estinction coefficeient、脂肪性索引、the protein’s grand average of hydrophobicity(GRAVY)value,和其他鹼性的physicochemical parameters,雖然這些似乎是非常簡單的程式,某一個可以開始推測關於蛋白質在細胞得所在地,例如,具有高度比例的lysine和arginine殘基的一個鹼性蛋白質,也許是一個DNA-binding protein。
PeptideMass(ExPASy)
設計用於peptide mapping的實驗,PeptideMass在暴露於protease,或是化學試劑之後決定一個protein的分裂產物,PeptideMass中可用於裂解的酵素和試劑為trypsin,chymotrypisn,LysC,cyanogen bromite,ArgC,AspN,和GluC(bicarbonate或phosphste),在合成peptide的分子量計算之前,Cystenines和methionines可以被修飾,藉由供應一個SWISS-PORT identifier而不是過去一排的序列,PeptideMass在SWISS-PORT註解之內可以利用知識去促進計算,例如,在裂解之前,移除signal sequences,或是包含已知的posttransnational modifications;在tabular format中給予開始的protein理論上的pI值及分子量,然後從SWISS-PORT來的變異中的the mass位置,被修飾的masses,以及peptide片段的序列,其結果可以被回復。
TGREASE
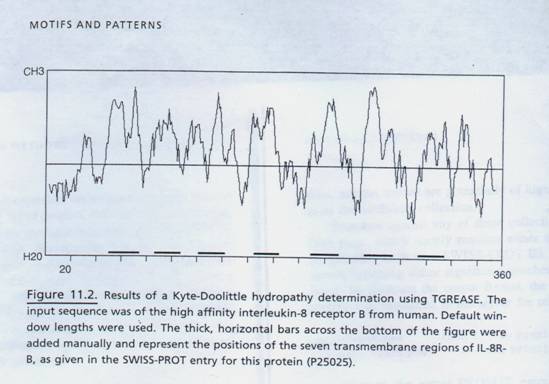
TGREASE順著protein的長度去計算它的厭水性,20個胺基酸天生具有它的厭水性:酸性的胺基酸相對傾向於陷入蛋白質的核心中,而遠離周圍的水分子,這個連結steric和其他considerstions的傾向,影響protein最終如何摺疊成其最後的3D立體結構,和球狀蛋白陷入區域之預測一樣,TGREASE在假定的transmembrane sequences可以找到應用,TGREASE是從University of Virginia得到之部分的FASTA suite程式,和像a stand-alone application一樣,可以被下載,而且可以跑Macintosh或DOS-based computers。
此方法依賴於hydropathy scale,每一個胺基酸基於很多的物理特性反應出其厭水數值(例如可溶性、經由蒸氣態轉移的自由能等等),胺基酸伴隨較高的正值為較厭水性的;較多負值的表示較親水性,移動的平均值或是hydropathic index可以穿過蛋白質而被計算,視窗的長度是可調整的,大約7~11個殘基的兼具,推薦minmize noise和masimize information content,這個結果的hydropathic index對於殘基的數目然後去作圖表,the human interleukin-8 receptor B的序列,被用來產生一個TGREASE圖表,如圖11.2顯示the peaks和the traesmembrane segments的實際位置間的一致性,雖然不是正確的,不過非常的好,要牢記在心這個分法,因為此方法可以預測所有厭水性的區域,不只是在這些位於traesmembrane區域而已,traesmembrane區域的特殊偵測法,下面會更進一步討論。
SAPS
The Statistical Analysis of Protein sequence(SAPS)運算法則對於任何給予查詢的序列提供廣泛統計學上的知識,當一個protein序列經由the SAPS Web介面,伺服器對於蛋白質可以回復成大量的物理與化學訊息,只能根據從序列來的暗示,輸出開始伴隨組成的分析,和胺基酸的總計,這個因循著電荷散布的分析包含正電或負電的位置、高數值的電荷及非 電荷的部分,和電荷的跑動,最後一部份和週期性分析一樣,在高數值厭水性和traesmembrane的部分顯示出訊息、重複的結構和multiplets。
MOTIFS AND PATTERNS
在第八章中,直接將序列比較的點子被呈現出來,那裡的BLAST searches在公開的資料庫中與所要查詢的序列類似可以執行去辨識序列;經常地,這個直接的比較也許不能產生想要的結果,或是一點也不能產生任何的結果。然而,有非常微弱的序列決定因素會呈現,來循著query sequence去和序列的family結合,藉由相同的步驟,一個序列的family可以用來辨識相同protein family之新的且關係疏遠的成員,這個的例子是PSI-BLAST(在第八章中討論),在討論利用這些的步驟的兩種方法之前,多數的條件,必須被定義,首先是輪廓的觀念,輪廓是十分簡單,是一個多樣sequence排列的數目描寫,大多像多數序列的排列,可從第九章討論出的方法得來,嵌入含多數序列排列內是本身序列的訊息,它代表帶有共有特徵且特別蒐集的序列,經常地為一個protein family,藉由輪廓可以運用這些嵌入且共同特徵,可以找出序列間的相似性,而這些序列鮮少有或沒有序列的一致性,要考慮到確認和疏遠關係蛋白質的分析,藉由拿一個多數序列排列代表的protein family使輪廓被構成,並且問了一連串的問題:
![]() 什麼樣的殘基可以在排列位置中被看到嗎?
什麼樣的殘基可以在排列位置中被看到嗎?
![]() 一個特殊的殘基多常出現在排列的位置嗎?
一個特殊的殘基多常出現在排列的位置嗎?
![]() 那裡有可以顯示完全保留的位置嗎?
那裡有可以顯示完全保留的位置嗎?
![]() 缺口可以在排列的任何地方被採用嗎?
缺口可以在排列的任何地方被採用嗎?
一但這些問題被回答,a position-specific(PSST)是可以被建構,而且在作業平台中的數目,現在可以代表多數序列的排列,多數包含PSST反映出任何既定胺基酸發生在每個位置的機率,也可以在排列時的每個位置反映出保守的或非保守的交替的效應,大多像PAM或BLOSUM matrix就是,這個PSST現在可以用來比喻抵抗單獨的序列,第二條件需要定義的是pattern或signature,這個signature通常也表現出protein family的共同特徵(或一個多數序列的排列),但是,不能包含任何重要的訊息,無論它為了某個殘基可以呈現在既定的位置而簡單地提供一套速記的記號,例如:the signature可以被讀成下列形式:
【IV】-G-X-G-T-【LIVMF】-X(2)-【GS】
第一個位置包含不是isoleucine就是valine,第二個位置只有glycine等等,X表示任何一個殘基可以出現在這個位置,X(2)簡單地意味兩個位置可以被任何胺基酸佔據,數字只是反映出沒有專一性的跑動。
ProfileScan
基於the classic Gribskov method of profile analysis,ProfileScan利用一個方法稱之pfscan,去找到一個蛋白質或核甘酸的query sequence和一個profile library間的相似之處,在這個事件有三個profile library可再搜尋時獲得,首先是PROSIDE為一個ExPASy database,可經由motif和序列輪廓的利用和patterns,將有生物學上有含意的位置去作成目錄,第二個是Pfam,它是一個protein domain families的集中,與多數如此蒐集方式在某個重要的觀點上有所不同:protein domains的開始排列,是藉由手完成的,而不是依賴自動照相機,本身地,Pfam包含稍許超過500 entries,但是這個entries可能有很高的特性,第三個profile set意指為Gribskov collection。
對於蒐集的搜尋,可以經由ProfileScan Web page去完成,它需要的不是在plain text format中輸入的序列就是identifier,如SWSS-PROT ID,使用者可以選擇搜尋的敏感度,回復只有重要的配對,或是所有的配對,其中包含一些不明確的案件,為了要解釋輸出的格式化,人類heat-shock-induced protein的序列被送至伺服器而只有準備PROSITE profiles的搜尋。
Normailized raw from-to Profile∣Description
355.9801 41556 pos. 6- 612 PF00012∣HSP70 Heat shock hsp70 proteins
雖然實際的PROSITE entry的回復,沒有很大的驚奇,輸出包含值得去理解的分數,未加以修飾的分數是實際的分數從搜尋期間scoring matrix而來的計算,較多訊息的數目是標準化或是N-score,N-score正式地代表配對的數目,此配對的數目在給予大小的database中被預期,在上述的例子中355的N-score譯成1.94×10-349期待機會配對,from和to的數字簡單地顯示query和matching的輪廓之間重複的位置。
BLOCKS
BLOCKS資料庫利用阻礙物的觀念去辨識一個蛋白質family,而不是依賴它們個體本身的序列,阻礙物的想法來自motif較親密的概念,通常起因於胺基酸的保存擴張,而此胺基酸可以給予蛋白質其功能或是結構,當這些個體motif來自於相同family中的蛋白質,其不需要引入裂縫而被排列時,結果是個阻礙物,而此”阻礙物”字義起因於排列,而不是他們本身序列的問題;明顯地,一個個體protein可以含有或多個阻礙物,且與它功能上以及結構上的motif一致。
BLOCKS資料庫可以從PROSITE中進入,當利用所要的序列去完成BLOCKS的搜尋時,query sequence在資料庫中所有可能的位置其排列不利於所有的阻礙物,為了每一個排列,分數的計算利用position-sepcific scoring matrix,而且最好配對結果回到使用者身上,搜尋可以隨意地抵抗PRINTS資料庫去執行,其中包含超過300 family的訊息不能進入BLOCKS資料庫,為了保證完整的適用範圍,它建議兩種資料庫都該被搜尋。
BLOCKS搜尋可利用在西雅圖內的the Fred Hutchinson Cancer Research Center中的BLOCKS Web site可以被執行,the Web site是容易的,允許用sequence或是關鍵字去執行,假如DNA sequence是用來作輸入,使用者可以記載基因密碼的使用和strand的搜尋,不管查詢是否經由序列或是經由關鍵字去執行,一個成功的搜尋將會回到到適當阻礙物,例如圖11.3,在這個entry(一個nuclear hormone receptor稱作steroid finger),它的首項按照順序標示著ID、AC和DE,藉由block family的簡短敘述、BLOCKS資料庫加入的數字和family的較長敘述而被描寫出來,Blline給予關於原始序列motife的訊息,使motife可以用來建構獨特的block,寬度和seqs參數顯示出block有多廣泛,在一些殘基中,和有多少的序列在block中,一些訊息循著關於統計學上的正確和結構的強度,最後,一個列表的序列被呈現,顯示出只有部分的序列和它獨特的motife有一致,每一條line會在序列上伴隨著加入SWSS-PROT的數字,第一個殘基的數字顯示基於完整的序列、序列本身,和position-based序列的重量,這些只可以用來製作成圖,伴隨著100個描寫中的序列,而此序列相較其他的group為最遠的,注意blank lines在一些序列之間,部分完全的排列是群集的而且在每一團有80﹪序列殘基是相同的。

CDD
近來,NCBI提供一個新的搜尋服務,目的在辨認含有蛋白質序列內保留的domain,這些搜尋的來源資料庫稱為the Conserved Domain Database或CDD,這是第二資料庫,會得到Pfam和SMART(Simple Modular Architecture Research Tool),SMART可用來辨識遺傳學上的domain和分析domain的結構,它會在第15章比較基因體學中詳細的比較,實際的搜尋利用reverse position-specific BLAST(RPS-BLAST)而被執行,RPS-BLAST利用查詢序列去搜尋一個計算過的PSSTs的資料庫。
CDD介面是簡單的,為了輸入的序列(兩者選一的,加入數字可以被記載),它提供一個盒子,並且為了選擇目標資料庫,而拉下明細表,假如保存的domains在輸入序列內被確認,以文字圖案顯示每個保留domain的位置,依循藉由查詢實際排列至目標domain,而由RPS-BLAST所產生的,在這些排列中,the default view顯示出相同的殘基是紅色的,假如所保留被替代則成藍色的,使用者也可以從牛血中的變異作選擇,其中包含典型的BLAST-style alignment display,Hyperlinks可以退回到源頭的資料庫,提供更多的訊息在獨特的domain,這個“CD Summary” page優先給予源頭資料庫訊息、參考資料、分類單位和在這群中具有代表性的序列,這個page比較低的部分中,使用者可以從這一群中建構想要的序列排列,兩者選其一地,使用者可以容許電腦去選擇高等級的序列或序列的子集合,這些在這群內很有分歧,如果3D結構和CD相符合可以被獲得的話,它可以直接用Cn3D看見,在CD連結可以進入CD Summary page,本質上地,從開始整個過程不斷的重複,運用序列去執行新的RPS-BLAST搜尋來對抗CDD。
SECONDARY STRUCTURE AND FOLDING CLASSES
在未知功能新發現的蛋白質或基因產物中,第一個步驟是執行BLAST或其他相似的搜尋來對抗公開化的資料庫,然而這些搜尋不能產生相符合的已知蛋白質;如果有統計學上的意義,在序列紀錄關於蛋白質二級的結構也許沒任何訊息,訊息在生化實驗的合理設計中非常重要,缺乏已知的訊息,必須有方法去預測序列行成α-helices及β-strand的能力,這個方法依賴於觀察一群的蛋白質,而此蛋白質的3D結構由實驗上決定,二級結構和摺疊類型的簡潔回顧,這他們本身的技術之前已經討論過了,暗示有厭水性side chain胺基酸的重要數字,那裡主要的chain或是骨架是親水性的,這兩個相反力之間是平衡的,可經由描述二級結構元素的組成而被達成,首先描述的是Linus Pauling and colleagues in 1951,α-helices是一個螺旋狀的結構,主要德chain形成骨架,而胺基酸的side chain則朝向helix的外面,骨架可藉由每個胺基酸的CO group和殘基四個位置的C-端(n+4)之NH group所形成的氫鍵來穩定,進而產生一個緊密、棒狀的結構,一些的殘基對於形成α-helices比其他來的要好,alanine、glutamine、leucine,和methionine通常可以在α-helices中找到,而proline、glycine、tyrosine,和serine通常是找不到的,proline通常視為helix的破壞者,因為它大量的環狀結構會破壞n+4氫鍵的形成。
相反的,β-strand為一個非常延伸的結構其穩定作用經由一個或多個鄰近的β-strand結合,而不是在此二級結構中形成氫鍵,經由這些個別的β-strand之間的交互作用所形成的完整結構稱之為β-pleated sheet,這些sheets可以平行或是反向平行,依賴於每個組成的β-strand其N-端和C-端的方向,β-sheet的變形為β-turn;在此結構中,polypeptide鏈製造出一個尖的,如髮夾彎曲,並且在次過程中產生反向平行β-sheet。
在 1976年,Levitt和Chothia提出一個分類系統,其基於蛋白質內的二級結構要素,十分簡單地,α-structure初期是由α-helices所形成的,而β-structure初期是由β-strand所形成的,Myoglobin為一個α-helices所組成蛋白質的典型例子,Plastocyanin是一個βclass的很好例子,氫鍵在八個β-strand間形成一個致密且為桶狀的結構;結合的類型,α/β,為先前的β-strand交替著α-helices所組成的,Flavodoxin為α/β的一個好例子,其中間為β-strand構成的中心,而周圍是α-helices。
從線形的初級的序列來預測正確二級結構的方式可以廣泛利用neural networks,其利用模式和傾向所作的分析,主要地,neural networks提供一個計算的過程,每個neural networks有輸入層和輸出層,在二級結構的預測事件中,輸入層為從本身序列來的訊息,而輸出層為獨特的殘基是否可以形成特殊的結構的機率,在輸入層和輸出層之間有一個或是數個隱藏層,實際的學習就是在那裡發生的,這可以藉由網路提供一個訓練用的資料而去完成,這裡,適當的訓練,則所有的序列之三度空間結構可以被推論出來,network可以處理訊息而找到胺基酸序列和結構可能的微弱關係,而可以形成一個特殊的背景,更多完整的neural networks應用於二級結構的預測,可以在Kneller等中找到。
nnpredict
nnpredict的運算規則是利用兩層、feed-forward neural network去分配每個殘基的預測型態,在做預測中,和蛋白質的folding class一樣(α,β或是α/β),伺服器可利用FASTA format和序列在單字母或是三字母中的密碼,殘基被分類為在α-helix(H),β-strand(E),或是都沒有,如果沒有預測可以作為記載,那一個question mark(?)可以被回復去指出工作無法執行,如果沒有關於folding class訊息可以利用,預測可以在沒有被記載的folding class而去做;這為default,為了best-case預測,nnpredict的正確率在報告中超過65%。
可以藉由送電子郵件留言到nnpredict@celeste.ucsf.edu或是藉由Web-based將序列送到nnpredict,用flavodoxin當做例子,電子郵件留言的格式化舉例如下:
option:a/b
>flavodoxin – Anacystis nidulans
PredictProtein AKIGLFYGTQTGVTQTIAESIQQEFGGESIVDLNDIANADASDLNAYDYLIIGCPTWNVGELQSDWEGIY
DDLDSVNFQGKKVAYFGAGDQVGYSDNFQDAMGILEEKISSLGSQTVGYWPIEGYDFNESKAVRNNQFVG
LAIDEDNQPDLTKNRIKTWVSQLKSEFGL
PredictProtein
PredictProtein用少許不同的方法去做預測,首先蛋白質序列在SWISS-PROT中查詢找到相似的序列,當相似的序列被找到,稱為MaxHom的運算法則可以用來產生profile-based多數序列的排列:在SWISS-PROT的首先搜尋之後,全部找到的序列用查詢序列和輪廓的計算來排列,輪廓被再次用來搜尋SWISS-PROT去設至於新的、配對的序列,MaxHom所產生的多數排列藉由PHD的方式供給到neural network去做預測,PHDsec,可以用來作為預測二級結構的方法,不分派每一個殘基到二級結構的類型中,而且在序列上的每一個位置中,提出統計學上的數值去指出預測的信用程度,這個方法產生平均的正確性大於72%,最好的殘基預測有超過90%的正確性,可以藉由送電子郵件或是利用Web front end將序列送至PredictProtein,多數送出去的序列是能利用的,查詢序列要當作單一字母的胺基酸密碼或是藉由它的SWISS-PROT identifier而被送出去,此外,在FASTA format中的多數序列排列或是如PIR排列可以因二級結構的預測而被送出去。
輸入的留言,寄至predictprotein@embl-heidelberg.de,如下列的型式:
Joe Buzzcut
National Human Genome Research Institute, NIH
buzzcut@baldguys.org
do NOT align
#FASTA list homeodomain protein
>ANTP
---KRGRQTYTRYQTLELEKEFHFNRYLTRRRRIEIAHALSLTERQIKIWFQNRRMKWKK
>HDD
MDEKRPRTAFSSEQLARLKREFNENRYLTERRRQQLSSELGLNEAQIKIWFQNKRAKIKK
>DLX
-KIRKPRTIYSSLQLQALNHRFQQTQYLALPERAELAASLGLTQTQVKIWFQNKRSKFKK
>FTT
---RKRRVLFSQAQVYELERRFKQQKYLSAAPEREHLASMIHLTPTQVKIWFQNHRYKMKR
>Pax6
--LQRNRTSFTQEQIEALEKEFERTHYPDVFARERLAAKIDLPEARIQVWFSNRRAKWRR
上面是homeodomain蛋白質為了要做二級結構預測的FASTA-formatted多數序列排列之例子,在名字、affiliation和住址後,#字的訊號至伺服器其序列為一連串單一字母的密碼,為了這個的排列,必須一開始有#字的訊號可以防止不會再重新排列,用電子郵件的方式將輸出寄出需要大量適切的訊息,從ftp site中結果可以藉由開始有#字的訊號之前,添加不能回信的限制者而被恢復,這對於這些電子郵件的伺服器是個有用的特徵,而此電子郵件不能處理大量的輸出檔案,輸出檔案的格式化可以是有或無的PHD文字圖形的plain text或是HTML檔案。
MaxHom搜尋的結果可以被回復,多數的排列可以用於更進一步的研究,如profile搜尋或是phylogenetic studies,如果送出的序列在PDB中有了相同已知的相同物,這個PDB identifier會被供給,在最近的釋放中,輸出也可以藉由記載可利用的選擇而被定做,PredictProtein回覆預測的可靠性之範圍為0到9,9為對於此二級結構的最大信用程度,也就是說此二級結構排列是正確地製作出來,當比較藉由其他方法獲得的結果,獨特的序列藉由伺服器可以回復結果,如圖11.4顯示出的修飾形式。

PREDATOR
PREDATTOR二級結構預測的運算法則以在胺基酸序列中可能地氫鍵殘基的認地作為根據,在不同類的局部氫鍵結構中,利用衍生自資料庫的統計學於residue-type的發生,這個方法的新奇特徵是對於序列局部的配對排列的信任,而可以在每個有關連的序列上去做預測,對於這個程式的輸入可以為單依序列或是一套無排列但有關連的序列,可以藉由predator@embl-heidelberg.de,或是藉由利用Web front end將序列送至PREDATOR的伺服器上,輸入的序列可以是FASTA、MSF,或是CLUSTAL format,在這三個結構狀態中,PREDATOR的平均預測正確率對於單一序列為68%,而對於一套有關連的序列而言則為75%。
PSIPRED
PSIPRED的方法發展於University of Warick,UK,利用從輸入序列的PSI-BLAST搜尋推論的知識去執行預測,PSIPRED利用兩個feedforward neural network去執行從PSIPRED而來的輪廓分析,序列不是在single-letter raw format就是在FASTA format中經由簡單的Web front end被送出,從PSIPRED預測的結果在電子郵件的留言中可以當作原文檔案來被答覆,除此之外,連結在電子郵件的留言中也可以提供二級結構預測的文字圖解描述,想像用Java的應用稱之為PSIPRED view,在這個描述中,helices和strands的位置可以在目標序列之上用圖表的方式去呈現,PSIPRED在結構上平均的預測正確率為76.5%,高過於其他被描述的方法。
SOPMA
The Protein Sequence Analysis伺服器在法國和Lyons裡的the Centre National de la Recherche Scientifique(CNRS)中,唯一在做二級結構的預測的步驟:不是用單一的方法,而是用五個,而達到consensus prediction,這個方法的利用為Garnier-Gibrat-Roban(GOR)方法、Levin homolog method、the double-prediction method、the PHD method描述部分PredictProtein,和CNRS本身的方法,稱為SOPMA,簡要地,這self-optimized預測方法用已知的蛋白質來建立蛋白質序列的次資料庫,每一個蛋白質在次資料庫中,是被投入為二級結構的預測,此預測基於序列上的相似性,從次資料庫來的訊息使在查詢序列上產生預測。
跑這個方法可以藉由在single-letter fomat中送本身的序列致deleage@ibcp.fr,利用SOPMA當作要投入的電子郵件留言,或是利用SOPMA Web介面,從每一個組成的預測來的輸出和consensus一樣顯示在圖11.4。
Comparison of Methods
在圖11.4的基本原理中,上面所有描寫的方式是好的,但不是完美的預測二級結構的工作,那裡沒有其他的訊息可以被知道,其最好完成的方法是利用運算法則判定預測的效力,並和其他方式做比較,Flavodoxin當作輸入的查詢而被選擇,因為它有相當錯綜複雜的結構,屬於α/β一類,包含六個α-helices和五個β-sheets,有一些排列藉由所有的方式一致性地被做出來,所有的方式公平地偵測β1、β3、β4和α5,然而,有一些方式完全的失去一些元素(如,α2、α3、α5的nnpredict),而有一些的預測製作出沒有生物學上的感覺(如double-prediction和β4,那裡的helices、sheets,以及殘基和殘基的互相交替),兩者都可以正確地找到所有的二級結構的元素,並且在數個可以辨識正確的結構的地方,呈現出已經做出最完整的預測,這不是說其他的方法沒用或是不好,無疑地,在一些例子中,其他的方法也可以當作很好的預測,這個方法不提供預測的錯誤保護方法,但是它可以增強預測結果的信用程度。
一個新的Web-based伺服器,Jpred,由六個預測的方式所組成,而且以主要的規則為根據去回復一致性的預測,這個伺服器的有效性因六個預測的運算法則而自動地產生輸入以及輸出,當要處理巨大的資料時,這是一個很重要的特徵;Jpred的輸入序列可以為單一的序列在FASTA或是PIR format、一套無排列的序列在PIR format,或是多數序列排列在MSF或是BLC format,在單一序列的例子中,伺服器首先藉由運用BLASTP運算法則的OWL資料庫搜尋,產生出一套有關連的序列,這個序列經SCANPS過濾,然後經AMPS做配對的比擬,最後成團的sequence set用75%的cutoff value去移除此sequence set的偏差,而且用CLUSTAL W將剩餘序列去做排列,Jpred伺服器跑PHD、DSC、NNSSP、PREDATOR、ZPRED,和MULPRED,從Jpred伺服器而來的結果可以用text file在電子郵件中回覆;連結在HTML或是PostScript file format中,也提供一個可以看見顏色的文字圖解描述,從Jpred伺服器來的一致性的預測,在三個結構狀態中,有72.9%的正確性。
SPECIALIZED STRUCTURES OR FEATURE
當α-helices和β-sheets的位置可以被以相對高程度的信賴度加以預測,存在一些深入的蛋白質構造或是特色可以被加以預測,例如:coiled coils和transmemb- rane regions。而在此並沒有像預測secondary structure那樣多的預測方法,主要是因為folding的規則誘使這些構造沒有完全地被了解。而當query sequences和已知結構的database所對應時,則預測的準確度就會很高。
Coiled coils
實行COILS algorithm是以query sequences和一個具有coiled coil結構的蛋白質的database去比對。而這種程式同時也在 query sequences和PDB的子集之間作比較,而確定哪一段sequence可以形成coiled coil。COILS可以VAX/VMS或者是利用簡單的網路介面加以下載。
COILS這種程式以GCC或是FASTA格式來得到sequence data,一段或者是多段序列必須馬上遵從。除了序列之外,使用者可以選擇兩種其中之一的scoring matr-ices:MTK,依據myosin、tropomyosin、keratin的序列。或者是MTIDK,依據myosin、tropomyosin、intermediate filaments types I-V、desmosomal protein、keratin的序列。作者引用一半的機會在兩種scoring matrices之間,以MTK去偵測two-strand structure為較佳,而以MTIDK去偵測其他全部的案件為佳。使用者可以利用選項去給在每一個coil的 a和d位置的residues(通常為厭水性部分)相同的重量,如同給b、c、e、f、g位置的residues(通常為親水性部分)。如果說COILS實行的結果顯現出有重量和無重量都相當地不同,則這可能是錯誤的正級被找到了,作者警告說COILS是為了去偵測solvent-exposed,left-handed coiled-coils所設計出來的,而buried或者是right-handed coiled coils則沒辦法去加以偵測。當 query sequences遵從Web server後,預測的圖表就會順著序列的長度而顯現出傾向於行程何種coiled coil。
一種較簡單去翻譯輸出物是由MacStripe而來,MacStripe為基於Macintosh理論的一種應用,利用Lupas COILS理論而去進行預測。MacStripe由FASTA,PIR,和其他共同檔案格式形成的coiled coil去得到input file,產生出來的圖表檔案包括:可能生成coiled coil 的histogram、heptad repeat pattern。
Transmembrane Regions
在之前被討論的Kyte-Doolittle TGREASE在偵測高厭水性區域時是一種非常有用的方法,但是他並非是獨有地預測transmembrane region,因為buried domains在可溶的、球狀蛋白質為主要的厭水性區域。我們認為第一要預測的方法是用來預測transmembrane regions。而這種方法為TMpred,依靠一種稱作為TMbase的transmembrane protein database,而TMbase是從SWISS-PROT所衍生出來的,包 括關於每段序列具有多少數目的transmembrane domain、這些domain的位置、及在自然狀況下在旁的序列有哪些。Tmpred利用這資訊和一些weight matrices加以連結而用以作預測。
Tmpred Web interface是非常簡單的。序列以one-letter code通過並進入query sequences box,使用者可以指定厭水部分transmembrane helix的最大及最小長度去加以進行分析。而輸出物總共包括4個部分:一個可能的transmembrane helices目錄、相關圖表、transmembrane 型態的建議模式、及一個以圖表描述的相同結果。
PHDtopology
PHDtopology在預測transmembrane helices時為其中一種有用的方法,而此種方法和之前討論過預測二級結構的方法-PredictProtein很類似。在這裡這種程式現在被用來觀察不一樣的方法去加以預測membrane-bound而非可溶性蛋白質。這種方法在預測上非常準確、幾乎完美的預測:預測transmembrane helix有92﹪的準確度,預測loop有96﹪的準確度,而同時預測以上2種情況有94.7﹪的準確度。而這種程式的特徵之一為除了預測假想的transmembrane regions,它還可以指出loop-regions相對於membrane的方位。
Signal Peptides
在丹麥的技術學院的生物序列分析中心發展出一套強力的偵測技術,而此種技術是在偵測signal peptides及其切割點,稱為SingnalP。此種技術是基於網際網路運作,使用已知的訊息序列當作training set去分離革藍氏陽性菌、革蘭氏陰性菌及真核細胞序列。SignalP預測非包括在細胞間訊息傳導的secretory signal peptides。
Nonglobular Regions
SEG可以被用來當作偵測蛋白質序列想像的nonglobular regions,而這種偵測方法是藉由改變trigger window length W、trigger complexity K1、extension complexity K2。當接受到seg sequence.txt45 3.4 3.75的指令時,SEG將會使用一段比長的window length,因此偵測到長的、nonglobular domains。
TERTIARY STRUCTURE
最複雜且在技術上要求嚴格的預測方法是基於蛋白質序列資料用以作為結構的預測。能夠成為適當的且準確的預測結構是基於對於序列的了解,而序列也許指定一種結構的形狀,而同樣的結構形狀也許可以被許多其他的序列所指定。結構被保存至較序列高的程度及有限度的back-bone motifs這個想法指出了在相似的蛋白質當中,透過慣例的、只以序列為基本的方法也許不是必須被偵測到的。推論序列跟構造之間的關係是protein-folding problem的根基,且目前在這個問題上的搜尋已經集中焦點在一些過去的評論文章。
在預測構造上最有力的技術是threading,而這種方法搜尋那些在folding時相似但是卻沒有明顯序列上相似的構造。這個方法抓到那些構造不輕的query sequ- ences,並且以在構造上已解決的對等的目標蛋白質穿過。序列在構造上可以從一個位置移動到另外一個位置,受至於一些預先決定的物理因素的拘束。例如:二及結構成分的長度和loop regions也許是固定的或者是具有許多不同的範圍。每一段序列對應構造的安置、pairwise和nonlocal residues間厭水性作用力是可被確定的。這種熱力學計算方式的程式是集中的計算結果、需求量最小的、為一個有力的UNIX workstation;且它們通常需要特別的電腦語言的知識。在以序列為基本的預測當中出現無法辨別結構時, threading methods是很有用的。
雖然threading methods這種技術明顯的是很有用的,他們現在的需求在硬體及專家見解上也許證明了對於生物學者而言是一種障礙。再試圖將障礙的高度變低時,easy-to-use這種程式已經被發展出來,並給予一般的生物學家對於比較蛋白質的模型化一個好的近似值。
SWISS-MODEL這種程式可以自動執行序列跟構造的比對,而這種程式是有兩個步驟的程序。First Approach mode是用來確定序列是否可以全部模型化,在一段序列被遵從,SWISS-MODEL將其與crystallographic database做比較。如果first approach 在ExPdb中找到一個或多個適當的記載,自動的模型將會被建築出來且將會使用最小的能量去生成最佳的模型。
另外一種自動蛋白質摺疊辨識方法在UCLA中被發展出來,一起預測二級結構的資訊,而這訊息是在探針序列上除了以序列為基本的配對至指定可能蛋白質摺疊到query sequences。在這個方法中,改正指派的摺疊是依靠探針序列所生成的ranked scores,是和以其在標的的三度空間結構的library中每一個構造的相容性為基準。
Second approach是在構造之間作比較。DALI在兩個蛋白質當中尋找相同成分的樣式,執行最佳的,並且對此蛋白質回溯最好的解決辦法。這種方法在缺口中視具有伸縮性的,且它讓調整部分之間交替的連接,因此促進辨識在兩種不同蛋白質之間相似的特別的domain,甚至是蛋白質之間都無相同之處也可以使用這種方法。DALI網路介面將會在兩個相對於PDB之間的部分執行分析的功能。如果感興趣的蛋白質都出現在PDB當中,他們預估的結構的鄰近物可以藉由FSSP database來找到,而FSSP是一種all-against-all去比較PDB的紀錄。
在這裡討論的最後一種方法擴大到之前討論到的PHD二及結構預測方法。TOPITS這種方法是一種可搜尋database,藉由在PDB中蛋白質轉錄的3-D結構到二級結構的1-D strings。然後二級結構和query sequences的可進入的溶劑藉由PHD加以確認,計算的結果也被紀錄在1-D string。之後quency和target strings被動力學的程式所加以調整,之後對構造加以預測。
在這裡所探討的方法確實是基本的,因此回報結果和它適應Web-style介面的能力都很迅速。它在偵測兩個蛋白質中較弱的結構的執行程度給人印象深刻。雖然蛋白質摺疊的問題還沒被完全解決,但是極多數蛋白質的摺疊可以藉由這些難以理解的方法去加以鑑定及信賴。因為不同的方法具有不同的張力及強度,通常是謹慎的使用consensus approach,和使用在預測二級結構的方法相似且較簡單。這些計算發展的時機的確令人相當興奮,由於同時間人類基因體計劃的完成,給予研究人員在假想的基因產物被鑑定出來時一個有力的處理方式去預測結構及功能之間的關係。
1. 首先由http://pdb.life.nthu.edu.tw/ 進入到清華大學的PDB鏡像站。(如圖一所示)
圖一.
![]()
![]()
![]()
2. 連接到蛋白質資料庫(Protein Data Bank)。
3. 若是知道PDB的code,可直接輸入code查詢。若是不知道code 可選擇 ”3DB Browser”(如圖二所示)
圖二.
![]()
4. 在keyword處鍵入=> 〝hydrogenase〞
5. 輸入完後,點擊〝Search〞
6. 將可得到11筆資料。
7. 選擇所要的資料,例如第一筆(如圖三所示)。
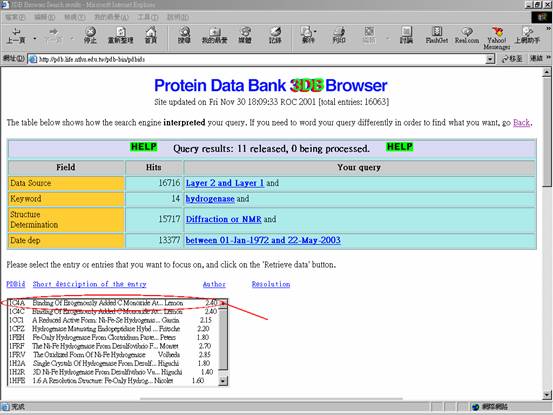
圖三.

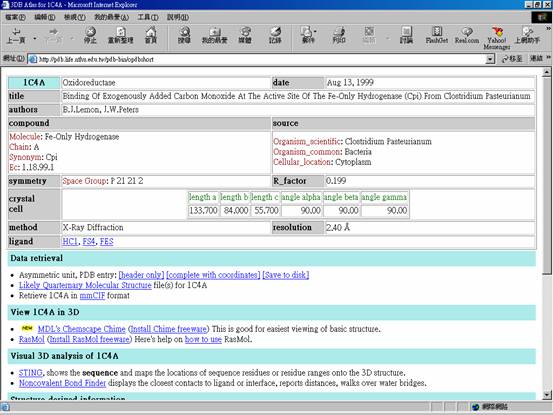
8. 網頁下拉,點擊Retrieve data(圖四)。將可得到詳細的資料(如圖五)。
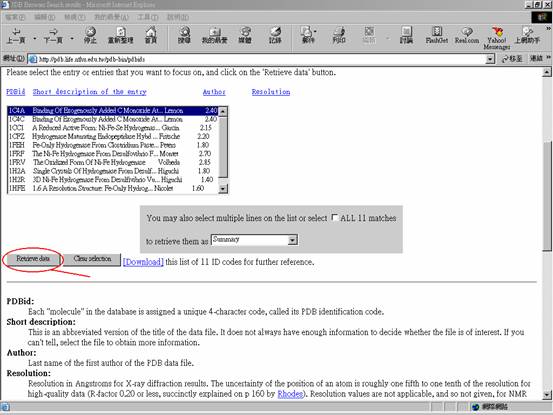
以下為詳細資料:
9.如果我們要看蛋白質3D結構圖,可利用分子視覺化軟體(Molecular Structure Visualization),在此利用Rasmol做介紹,可至網站http://www.umass.edu/microbio/rasmol 下載。
或下載頁面:
http://www.umass.edu/microbio/rasmol/distrib/rw32b2a.exe下載。
1.按file讀取檔案
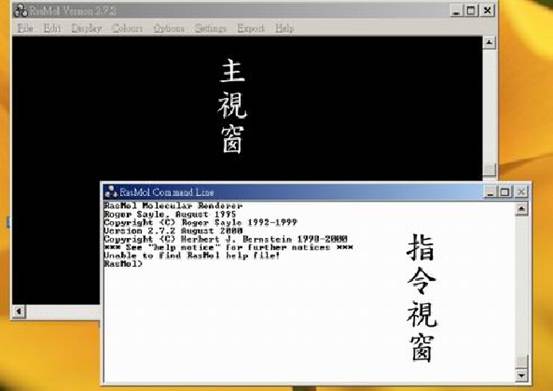
主視窗: 顯示結構的視窗。
指令視窗: 鍵入指令的位置。
2.如何觀看蛋白質結構有多少鏈結?
 輸入下列的指令到指令視窗當中
輸入下列的指令到指令視窗當中
指令:
reset
rotate z 90
zoom 150
rotate y 40
Display -> backbone (on)
color chain
點擊每一個鏈(chain)就會呈現出它們身份証字號(ID letter code)(如上圖)
show sequence
這一個指令將呈現出完整的胺基酸序列和有關於鏈(chains)的一些資訊.
3. protein/DNA 之外的物質
在PDB file 中,除了protein/DNA 鏈chains 之外,有沒有其它的東西?
指令:
select hetero
選擇所要的東西,(選擇除了蛋白質、DNA 和RNA 之外的東西。)。如水、金屬離
 子或其它物質。.
子或其它物質。.
spacefill
color cpk
restrict not water
在此不把水顯現出來。在舊的PDB
檔案中有一些模糊的
定義。例如CD
可以代表
delta 碳或鈣。
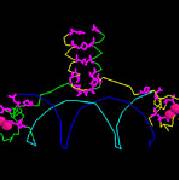
4.如何呈現疏水性的胺基酸?
指令:
select hydrophobic
color magenta
wireframe 0.4
注意: alpha 螺旋是兩性分子。
select not water
spacefill (on)
slab (on)
此圖可以看出分子內部疏水性和親水性殘基的分部。
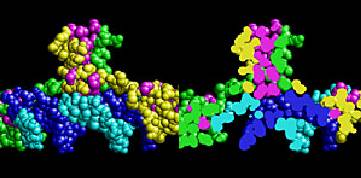 利用滑鼠移動平板面(
slab plane)
利用滑鼠移動平板面(
slab plane)
5. 如何呈現Cd 離子的位置?
指令:
slab off
select all
Display -> backbond
backbone 0.3
 color
chain
color
chain
select cd
spacefill (on)
color cpk
select within(2.6, cd)
此指令是代表在距離Cd++ 2.6 埃內的所有原子。
spacefill on
color cpk
6. 如何儲存影像?
指令:
save script gal4.spt
zap
7. 如何執行script?
指令:
script gal4.spt
第六項是可以將所執行過的指令存成script 到此一項目執行
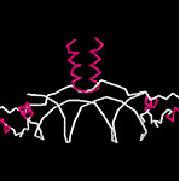 8.
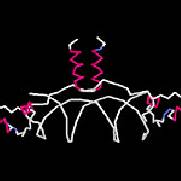
哪裡是a-螺旋和b-長帶?
8.
哪裡是a-螺旋和b-長帶?
指令:
Select all
Display -> backbond
Backbone (0.3)
color structure
紫色是a-螺旋、黃色是b-長帶(1d66.pdb 沒有b-長帶).
structure
color structure.
這是由RasMol 自己決定顏色。例如
二級結構是使用DSSP 的定義。
注意: 轉折(turns)是代表藍色。
9. 要如何找出兩個原子間的距離?
指令:
spacefill (on)
set picking distance
現在點擊兩個原子,看一看顯示在指令視窗的訊息。
假如你想要標示兩個原子和距離,試著鍵入:
set picking monitor
現在在分子的邊邊點擊兩個原子
(若背景為黑色,則顯現效果會很好喔。)
注意輸入下列指命的變化:
color monitor white
set monitor off
號碼將會不見!
monitor off
虛線將會不見喔!
set picking ident
上列指令是恢複正常的點擊功能,如此才可點擊每個原子並得到每個原子
的資訊。
RasMol 也可以呈現角度和立體角(torsion angles)。參照
www.umass.edu/microbio/rasmol/distrib/rasman.htm#setpicking
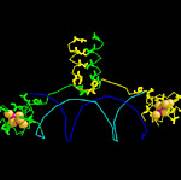
10. 要如何找出蛋白質和DNA 的鍵結?
指令:
reset
Backbone
color green
backbone 0
rotate z 91
translate y -17
zoom 200
select dna
color white
spacefill
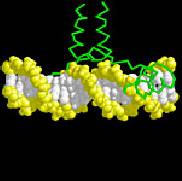 center
selected
center
selected
select dna and backbone
color yellow
現在你可以看到DNA
它的主鏈,主鏈(
backbone)和
鹼基對(base
pairs)呈現不同的顏色。你可以看到蛋白
質的主鏈是綠色的線喔!
select within(3.1, dna) and not dna
選擇距離DNA 3.1 埃內,但不選擇DNA! 上列的指命應該會選出35 個原子
喔!
color cpk
dots
現在按鍵盤上的"向上箭頭"直到出現你可以看見"裡面--select within(3.1,
dna) ......",然後加入and not water. 按下Enter,總共選擇了19 個原子。
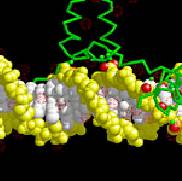 spacefill
0.6;
spacefill
0.6;
select within(3.1, protein) and dna
color cpk
現在你可以放大並點擊donor
和acceptor
的殘基,然後評估它們的氫鍵。
11. 要如何看到一個分子的內部?
指令:
在鍵入下列指令時,不要在任何時刻用滑鼠移動分子
reset
select All
spacefill
color Chain.
rotate x 83
zoom 200
set hetero off
關掉水分子
select dna
color cpk
slab
開啟平板面模式( slab mode)
在剖面之前的原子將看不到,只看到剖面之後的物質。
set slabmode section;
現在只有剖面圖呈現出來。在剖面的前面和後方的東西將不會呈現出來,
只有在"刀面的"原小才會展現。
slab 76
現在你可以看到GC 鹼基對, 可以看到三個華生-克里克的氫鍵
(Watson-Crick hydrogen bonds) (假如你用滑鼠移動了分子,你將會看不到就得要重設了。)
slab 68
這是什麼?利用滑鼠移動平板面(slab plane) (按住Ctrl、然後點擊、拖曳上和
下) Can 你是否可以發現有個鹼基對完全在華生-克里克位置(Watson-Crick
position) 之外? (答案在這份文件的最後面)
12.要如何要在固定DNA 下旋轉視窗?
指令:
reset
restrict none
Clear the screen
restrict dna
spacefill
rotate z 90
zoom 200
試著繞著DNA 螺旋的軸旋轉。(滑鼠上、下移動試試).
注意DNA 是繞著質心上下移動,所以未呈像的蛋白質也會跟著繞喔!
center selected
現在再試一次、注意有何變化
13. 對於相同的原子如何有各種不同的表示法?
指令:
restrict :d
color cpk
:d 代表在D 鏈上的所有原子.
backbone 1.
要確定在1 之後有沒有小數點,因為會讓Rasmol 認為單位是埃。
當在指令視窗下鍵入有關display 的指令時,現有的圖形表示將不會被關掉,
若在主視窗選擇display 的項目就會關掉現有的圖形。
"Sticks" are wireframe with a nonzero radius. Balls are spacefill with a uniform
radius.
spacefill off
wireframe 0.5
wireframe 0.1
spacefill 0.3
backbone 0.1
zoom 500
14. 要如何標示原子?
指令:
set picking label
現在點擊一些原子。
color labels white
label off
set picking ident
點擊原子並注意它的ID number 。它的號碼將視為下一個指令的### 。
select atomno = ###
label "My Favorite Atom"
label %n%r
標示出殘基的名字和號碼
label
標示所選擇原子的全部資訊。
label off
注意
對於"set picking" 指命的參數如下:
ident report an atom identity
distance report the distance between two atoms
angle report the bending angle defined by three atoms
torsion report the torsional angle defined by four atoms
monitor
Toggle the displayt of the measurement such as distance, bending or
torsional angles in the Main window
label Toggle the display of an atom label on a given atom
center set the atom picked as the center of rotation
可標示特定的項目:
%a Atom Name
%n Residue Name
%r Residue Number
%c Chain Identifier
%i Atom Serial Number
%e Element Atomic Symbol
15. 要如何看到分子的立體圖(stereo)?
指令:
stereo (on)
stereo -5
對於大部分的研究而言,此一步驟並非必要。在沒有立體圖的情形下,旋
轉(Rotation)將有助於了解3D 結構的關係。然而,假如你常常看分子圖形,
學習如何看立體圖將有助於你對於複合體空間結構的研究。Gale Rhodes 提
供了一個有關於立體圖的簡介( stereo
viewing)macweb.acs.usm.maine.edu/chemistry/GR/GraphicsGallery/StereoView.html
16. 如何指定特定的鏈、殘基、原子?
大部份在Rasmol 中,對於原子的描述指命如下:
lys45:a.nz
指出在鏈A 上第45 個位置上的lys 它的N zeta 原子
下表列出有關於原子的表示方法:
:a 列出在鏈A 的所有原子
lys:a 列出在鏈A 的所有lysine
45:a 列出在鏈A 的第45 個殘基
*45:a.nz
列出在鏈A 上第45 個殘基的N zeta 原子(注意假如45 號殘基的
名字不是明白的指定時,星號是一定要的)
17. Tips
在主視窗中,消除現有的展現圖形。鍵入
restrict none
18. 滑鼠

補充資料
· 蛋白質資料銀行(Protein Data Bank,PDB);
利用現有結構資料庫再對結構進行分類分析運算所衍生出的資料
庫:SCOP、CATH。
· 蛋白質3D 結構(PDB)
PDB 美國Brookhaven 國家實驗室管理的蛋白質資料庫銀行
使用核磁共振(Nuclear Magnetic Resonance, NMR)、x-ray 繞射實驗、理論模擬解出蛋白質的三度空間立體結構。
主要的資料有:原子空間座標、引用文獻、胺基酸序列、形成a-helix,
b-sheet 二級結構部份的胺基酸序列、雙硫鍵(disulfide bond)連結模式、參與生物功能的胺基酸殘基(residue)、與蛋白質結合的受體分子(ligand)。
·分子視覺化軟體
(Molecular Structure Visualization)
是一種可顯現出生物巨分子結構的軟體,包含蛋白質、DNA、RNA、化學小分
子和金屬等皆可藉由視覺化軟體來呈現他們的結構。
一般來說,視覺化的軟體可以輔助觀察巨分子的結構、作用力、表面特性等。尤
其在藥物設計、分子模擬上有很大的應用空間。
Graphical tools
RasMol http://www.umass.edu/microbio/rasmol/
Chemscape ChimeTM http://www.mdlchime.com/chime/
MolPOV http://www.chem.ufl.edu/~der/der_pov2.htm
MolMol http://www.mol.biol.ethz.ch/wuthrich/software/molmol/
Ribbons http://www.cmc.uab.edu/ribbons/
MolScript http://www.avatar.se/molscript/
WebLab ViewerLite and ViewerPro
http://www.accelrys.com/about/msi.html
Swiss-PDB Viewer http://www.expasy.ch/spdbv/
XtalView http://www.scripps.edu/pub/dem-web/toc.html
MolView and MolView Lite http://bilbo.bio.purdue.edu/~tom/
1. 清大生科系,呂平江教授授課講義。
2. PDB鏡像站http://pdb.life.nthu.edu.tw/
3. Rasmol網站http://www.umass.edu/microbio/rasmol
4. 呂平江教授授課pdf檔http://brc.se.fju.edu.tw/plans/slides/030502/0502pdf2.pdf