蛋白質的純化
口述:莊榮輝 老師
整理:林佳褀、郭裕民、黃保臻、呂昀
1. 簡單蛋白質:是指以酸、鹼或酵素水解後,只能產生胺基酸或其衍生物。如:球蛋白(蛋類的卵球蛋白、血液中的血清球蛋白)。
2. 複合蛋白質:是由單純蛋白質與非蛋白質物質結合而成。如:醣蛋白(蛋白質與醣類物質結合)。
3. 衍生蛋白質:是單純蛋白質或複合蛋白質之分解產物,這包括分子內的重新組合但未被破壞月生肽鍵者。如:多月生肽(Polypeptides)。
1. 纖維狀蛋白質:由眾多生太鏈以平行方式連接成直線狀,它們通常不溶於體液中,且具有張力、韌性;如:頭髮、指甲中的角蛋白等。
2. 球形蛋白質:由胺基酸之間的鍵成環狀且受緊密壓擠所形成之球狀或橢圓球狀,通常可溶於體液中,如血紅蛋白等。
要仔細研究一種蛋白質的結構與功能(structure and function),必須把此蛋白質純化出來。基本上,可利用蛋白質的一些特性,將其從細胞內無數的蛋白中分離出來。純化的方法一般是利用其溶解特性、色層分析特性和分子量來設計的。當這些方法都無效時,可採用更精細的方法。純化過程的每個階段,都必須測試酵素的專一活性,即每毫克蛋白質中酵素的活性,它顯示出酵素的純度。
傳統的純化方法是以較貴且較不經濟的鹽析法(salting out) 加鹽(通常為氯化鈉)於一有機固體之水溶液中,溶解之鹽離子會吸收並保有水分子,以減少水分子與溶質(即該有機固體)作用的機會,減低有機物對水之溶解度,而分離或沈澱出來。蛋白質、肥皂即類似物質之膠體浮液即以此法沈澱。
1、粗蛋白質︰採樣 → 均質打破細胞 → 抽出全蛋白,多使用鹽析沉澱法;可以粗略去除蛋白質以外的物質。
2、部分純化︰初步的純化,使用各種 管柱層析法。
3、均質酵素︰目標酵素的進一步精製純化,可用 製備式電泳 或 HPLC。
萃取的蛋白質,可以進行各種純化方法,例如硫酸銨沈澱、膠體過濾法、離子交換法、親和層析法等,以分離目標蛋白質現今所使用的酵素純化分離的方法,大致上可分為:
(1)鹽溶鹽析,此為普遍使用、且經濟方便的蛋白質純化分離方法
(2)膠體過濾,依照分子大小做篩選,而達到分離純化的目的
(3)離子交換,利用各分子間帶電荷量的不同來做篩選分析依據
(4)親和層析,利用不同結構的分子間的專一性結合來做分離純化
(5)最後的純化策略,是依照目標分子所擁有的特性,綜合使用各種純化分離方式,以達到最大的分離效率與純度。
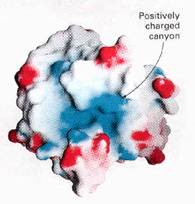

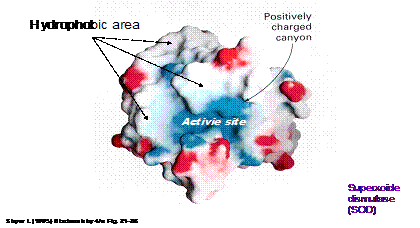
要純化一個酵素,或說是一個蛋白質,首先一定要瞭解這個分子的結構以及特性。就舉lysosome來說,我們要瞭解其組成骨架以及其親水、厭水區的分佈(如圖1),再者,要考慮分子上所攜帶側基的種類,是否帶有質子而使得分子帶電性偏向帶正電,亦或恰巧反之。
質子的存在,影響不僅周圍環境的酸鹼度,也同時影響到了一個分子其帶電性,而此時就要提到關於等電點(isoelectric point)的觀念了。所謂的等電點,就是一個分子其所帶的淨電荷為零時的pH值,此為一個分子的物理特性,而一個分子的等電點與其所處周遭環境的酸鹼值即直接地影響了這個分子的帶電性質,若周圍環境的pH值高於此分子的等電點,則此時分子帶負電,而反之則帶正電,此種分子特性不但影響到了其結構變化以及其所帶的酵素活性,亦可以此做為純化分離蛋白質的依據。
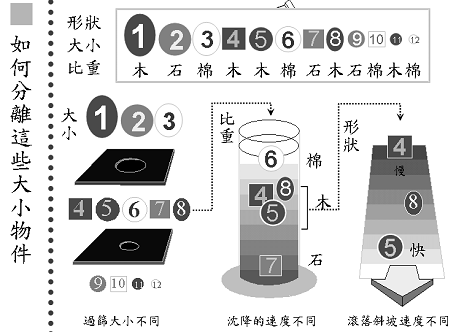
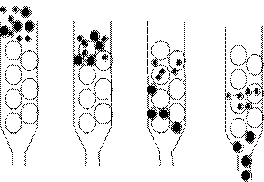
分離不同的蛋白質就像在分離不同特性的積木(圖2):假設現在要分離12個物體,有大有小,比重也不太一樣,有木頭、石頭、棉花,形狀也不太一樣。要從其中選出5號木球,要先想辦法偵測它,假設它會發亮,於是在每一次的分離作用中,就可以找出有發亮的那一部份逐一篩選。一開始先用大小做篩選,再利用沈降速度的不同,最後利用滾落斜坡速度不同,在每一次的分離方法中把並非我們所要的物質排除,最後挑出我們所要的5號木球。在作蛋白質純化時,原理也是一樣,利用蛋白質的不同性質,把所要的蛋白質從一堆物質中分離出來。
SHAPE \* MERGEFORMAT
圖2:可利用物質的不同特性,將所要的物質分離出來。
4.細胞中蛋白質的純化與分離(圖3)
要純化細胞中某一個酵素,第一件事要先把細胞作均質,把細胞打碎,打碎之後分成幾個部分,其中的細胞碎片是不要的部分,可溶的部分可分成小分子及巨分子,巨分子中有核酸、蛋白質、多醣和脂質(脂質實際上是小分子,但很容易聚集成大分子)。在巨分子中我們主要是要蛋白質,第一步用硫酸銨就可以把巨分子中的大部分蛋白質沉澱下來,核酸和多醣一般來說不容易被硫酸銨沈澱,所以在這一步就可以把大部分的核酸及多醣去掉,只留下蛋白質。這些蛋白質於是可以作進一步分離,可利用:
1. 分子大小不同,例如膠體過濾法
2. 分子電荷不同,例如離子交換法
3. 用分子極性不同,例如salting out
4. 親和力不同,例如MCAC (Metal Chelate Affinity Chromatography)
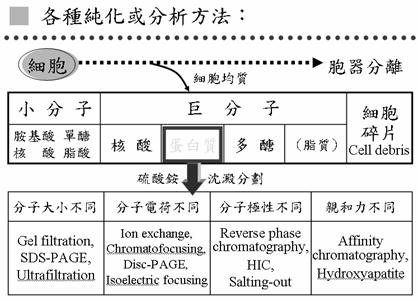
圖3:各種純化及分析蛋白質的方法
Ultrafiltration:超微過濾,利用一個帶有很多小洞的膜,其大小可辨別分子大小。
Chromatofocusing:現在較少用。
HIC:hydrophobic interaction chromatography,跟HPLC很像,但是是利用低壓。
Hydroxyapatite:一種陶土,可吸附蛋白質,吸附的原理尚不清楚,對不同的蛋白質有不同親和力。
我們所使用的鹽溶鹽析(salt in / salt out)亦包含了等電點的原理,鹽析沉澱法可分離出樣本中的蛋白質。蛋白質在水溶液中的溶解度,會受到溶液中的酸鹼度及離子濃度的影響而改變,可利用此特性來沉澱蛋白質。其原理是因為蛋白質分子表面電荷的改變,或者分子上極性或非極性區域與水分子間作用結果。
鹽溶(salt in) -當環境的pH值和其蛋白質的pI值相等時,蛋白質所帶的淨電荷為0,所以分子間的排斥力降低,而導致蛋白質分子之溶解度下降,進而聚集、沉澱,若在此時,提高了溶液的鹽類濃度(如:加入NaCL或KCL),則鹽類所解離出的正負離子會阻斷蛋白質分子之間正負電荷的相吸,因而使蛋白質的溶解度上升,此即為鹽溶現象。在環境酸鹼度離分子等電點越遠時,此鹽溶現象越加明顯。
鹽析(salt out)- 原理:在蛋白質分子表面的疏水性區域聚集著許多水分子,當我們把鹽類加入時,水分子便會脫離蛋白質表面,與鹽離子進行水合,而暴露出來原來表面的疏水性區域,進而相互結合形成較大的分子沉澱。主要經常利用加入硫酸銨(ammonium sulfate)來提高溶液中鹽類的濃度,因為硫酸銨所解離的離子容很大,所帶的電子數也多 (NH4+, SO42-),因此當其溶入水中時,會吸引大量水分子與其水合,造成水籠(clathrate)無法隔離非極性殊水區域,因此蛋白質的非極性與非極性分子會因親合力而互相吸引結合,進而聚集、沉澱,造成蛋白質沉澱出來,因而可用來分離蛋白質。
 至於水籠(圖4),也就是屬於疏水性的物質若將之置於水溶液中,水分子會聚集在非極性分子的四周,形成一個籠子般的結構,將非極性分子包圍在其結構骨架之中,以此種稱為水籠(clathrate)的方式來把非極性區域做隔離,而形成水籠的水分子其分子間氫鍵較穩定且分子本身的流動性也較低。
至於水籠(圖4),也就是屬於疏水性的物質若將之置於水溶液中,水分子會聚集在非極性分子的四周,形成一個籠子般的結構,將非極性分子包圍在其結構骨架之中,以此種稱為水籠(clathrate)的方式來把非極性區域做隔離,而形成水籠的水分子其分子間氫鍵較穩定且分子本身的流動性也較低。

在蛋白質的純化分離中,在採樣後先以超音波震盪或使用均質儀等方法將細胞打破以取出其所有的蛋白質,接下來第一步通常即以鹽析來做初步的純化分離,而得到的蛋白質則稱之為粗蛋白。
經過初步的分離後第二步通常則使用各種管柱層析法,而最後再以電泳或是HPLC來取得最終精製純化的均質酵素。而層析法在經過多年改革後,以從原先的使用濾紙做擴散分離,發展到今日使用管柱狀進行更大量的純化分析。色層分析法是由流動相與固定相所組合而成的一個系統,流動相與固定相則分別有不同的極性或非極性,而樣本分子若為極性則易於溶入兩相之中屬於極性的一相,反之亦然,此即為層析法的基本原理。而現今所使用的膠體過濾法(gel filtration)是一種分隔型的層析法,其固定相是由帶有細小孔洞的膠球所組成的,當樣本分子隨著流動相流過固定相時,小分子較大分子易於流進膠球的孔洞之中而較慢流出,依此原理即可以設計不同孔洞大小之膠球來分離不同分子大小的蛋白質,而要特別注意的地方,則是在裝填膠球時要盡可能地減少無效空間(dead volume),以增加純化分離的準確精細度。

要大量純化酵素時,通常前面的步驟還是要使用傳統層析方法,再加上HPLC或FPLC 則可以得到更純的均質蛋白質;製備式電泳 及超高速離心法,也可以增加純度,但處理量較少;而超微薄膜過濾法並非純化步驟,但在純化的各階段過程中,可濃縮蛋白質並除去小分子。

(a)膠體過濾的原理:
利用裝置於玻璃管柱內有孔膠體小珠(bead),依分子篩原理,當欲分離分子直徑大於珠孔直徑時,此分子會直接經由小珠間隙流出。當欲分離分子直徑小於珠孔直徑時,此分子會進入小珠孔內而延滯其流出時間。因此膠體過濾法可被用於測度物質之分子量大小,通常分子量大者分子直徑較大,流出速度較快;分子量小者分子直徑較小,流出速度較慢。
(b)膠體的來源:
一般常用高分子量多醣分子Sephadex,Sephadex是細菌合成的一種糊精(dextran),具三度空間網狀結構,將其製成顆粒狀使用。
(c)膠體的選擇:
依網目(mesh)大小來選擇,一般常用Sephadex G-10至 G-200的膠體顆粒,其英文字母G之後的數字越大者網目越大。
(d)大分子的分離:
DNA的分子很大,Sephadex G-10至 G-200的膠體顆粒均無法令DNA的分子進入膠體顆粒而分離。近年來已有大網目膠體顆粒的製造,如Sephadex G-1000可分離plasmid。另有Sephadex 4B、Sephadex 2B等大網目膠體顆粒的製造,其fractionation range分別達3×105~3×106與2×106~25×106 MW,可分離Virus等物。
(e)膠體過濾的優缺點:
就物質分離而言,膠體過濾法、薄層色層分析法與透析法均可分離物質,唯薄層色層分析法較適用於小分子物質之分離,而透析法的分離時間太長。超大物質的分離則不適於利用膠體過濾法,膠體過濾法的另一缺點為需大量的sample。近年來發展另一種分離工具微量離心過濾膜,藉離心過濾法分離各種分子大小物質,並可快速分離。就物質分子量的鑑定而言,膠體過濾法、電泳法與超高速離心法均可鑑定分子量大小,唯電泳法費時,而超高速離心法則費時、費錢。
1.離子交換樹脂
 蛋白質上會有帶正電和帶負電的基團,因此可利用這些帶正電和帶負電的基團,以及蛋白質等電點的性質來進行離子交換樹脂(圖7)。環境中的pH值會影響一個蛋白質的靜電荷(net
charge);舉例來說,一個pI值為6的蛋白質,若將其環境的pH調到7,則此蛋白質的靜電荷將為負,所以就用陰離子交換法(anion
exchange)。若把環境的pH值調到5,這時蛋白質的靜電荷將會為正,因此要用陽離子交換樹脂(cation
exchange)。
蛋白質上會有帶正電和帶負電的基團,因此可利用這些帶正電和帶負電的基團,以及蛋白質等電點的性質來進行離子交換樹脂(圖7)。環境中的pH值會影響一個蛋白質的靜電荷(net
charge);舉例來說,一個pI值為6的蛋白質,若將其環境的pH調到7,則此蛋白質的靜電荷將為負,所以就用陰離子交換法(anion
exchange)。若把環境的pH值調到5,這時蛋白質的靜電荷將會為正,因此要用陽離子交換樹脂(cation
exchange)。
圖7:蛋白質表面有許多帶有正電或負電的離子基團,可供離子交換之用
若把環境的pH值調到9,仍然是用陰離子交換樹脂,但這和環境的pH為7時,情況是有差別的。當環境是pH 7時蛋白質是帶負電,環境是pH 9時蛋白質也是帶負電,但環境的pH值越高時,這個蛋白質帶的負電也越多;假設蛋白質在pH 7時是帶一個負電,在pH 9時是帶三個負電,帶三個負電的蛋白質吸附樹脂的強度比帶一個負電的蛋白質來的強;如此一來,最後要把所要的蛋白質溶離出來時,就要使用相對更強的環境,才能破壞蛋白質和樹脂間的結合力。因此在使用離子交換樹脂時,要注意不要讓蛋白質帶太強的負電或正電,因為蛋白質若帶太強的電荷,就必須要用很強的環境才能把此蛋白質溶離下來,這樣一來就很可能會破壞到蛋白質的活性;一般的原則是將環境的pH值調到比蛋白質的pI高一個pH或低一個pH就可以了。
以陰離子交換法做一個例子:用陰離子交換樹脂就是要分離純化帶陰離子的樣本分子,則用來吸附它的擔體(固定相)就要帶正電荷。離子交換法也是一種色析法,因此也有所謂的流動相及固定相,固定相即為擔體,在此例子中帶正電;流動相則是樣本及
緩衝液(buffer),其中帶陰離子的分子就會吸附到擔體上,不帶電或帶正電的分子,就會被溶離下來。陰離子被吸附的程度則由它所帶的電荷決定。吸附到擔體上的負電樣本分子就叫做counter ion。Counter ion被吸附到擔體上之後,要再將蛋白質從擔體分離出來時,一般來說都是用鹽梯度,因為陰離子和陽離子間是以離子鍵結合,可以用NaCl來破壞這種吸引力,其中Na+會跟負電結合,Cl-則會跟正電結合。
假設現在要把帶兩個負電及帶一個負電的蛋白質從這個擔體溶離下來,漸漸提高鹽的濃度,則帶一個負電的會先被溶離下來,接著是帶兩個負電的蛋白質。要注意的是,有的蛋白質無法忍受太高濃度的鹽,可能會導致蛋白質變性,失去活性。
離子取代的順序叫pecking order,在離子交換法中,離子交換的順序原則是電荷高的取代電荷低的,若是同電荷則離子大的取代離子小的,濃度高的會取代濃度低的。因此我們在作離子交換樹脂時,讓樣本吸附上去之後,再利用濃度的差異,提高某一種溶離液的濃度,把蛋白質一個一個沖洗下來。除了利用NaCl之外還可以利用pH值的變化,將pH值漸漸下降,因此質子的濃度會漸漸提高。
拉鹽梯度時有兩種方式(圖8),一種叫連續梯度,一種是階段梯度,兩種方法各有特點。在拉連續低度時,需要有梯度製造器;而階段梯度則是過一段時間換一種濃度的溶液。在連續梯度的方法中,舉例來說,若要從0M拉到1M的鹽梯度,就在低限溶液中放Buffer,高限溶液中則放1M的鹽和Buffer;高限溶液和低限溶液中有通道,低限溶液有另一個出口可讓溶液出來到管柱,因此一開始出來的都是0M的溶液,由於連通管原理,從低限溶液流出來多少溶液,高限溶液就會補進多少溶液,在低限溶液這邊有一個攪拌器,可以將其攪拌均勻,因此流出的鹽濃度會漸漸提高。
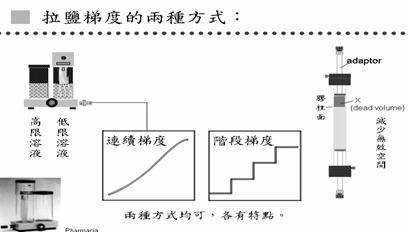
圖8: 拉鹽梯度有兩種方式,連續梯度需用梯度製造器,階段梯度則每過一段時間換一種濃度。
在整個離子交換法中很重要的一點是,真正有分離作用的只有膠體的部分,在膠體上方的空間叫無效空間(dead volume) ,在作離子交換法時,要盡量減少無效空間,因為無效空間會使得流進來的濃度梯度被破壞,失去梯度的連續性,作出的實驗結果就很差。有的管柱附有adaptor,可利用adaptor盡量減少無效空間。
2.親和層析法
親和層析法(圖9)的純化效果很好,但不是每一種蛋白質都可以利用。同樣的,在親和層析法中也有固相的擔體,跟離子交換法和膠體過濾法一樣,擔體都是很小的珠子,這些珠子上可以接上某些分子,任何可以抓到所要物質的分子都可以接到珠子上。但在親和層析法中,一定要先找到有親和性配對的分子,例如:抗體和抗原,酵素和基質,荷爾蒙和接受器。最常利用的是抗體和抗原,若將抗體接到珠子上然後讓樣本流過,則抗體就會將其中所含的抗原抓出來,其他不要的分子就會被沖洗下來,接下來就可以把所要的物質溶離下來(親和層析法的缺點是常要用很強的力量把所要的物質溶離下來)。
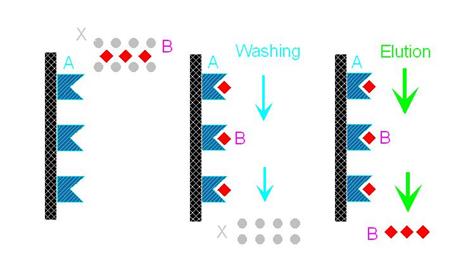
圖9: 親和層析法的作用機制-
圖中的A可視作抗體,B則是其抗原,X則為不要的雜質。在第一個圖中,B夾雜在樣品中;第二個圖中,B和A吸附在一起,雜質則被沖洗出來;第三個圖中,B就被溶離純化出來了。
親和層析法的先決條件要找到有親和力配對的物質,並不是每個蛋白質都可以找到,但現在遺傳工程的技術可以在蛋白質序列的前面或後面加上一個tag,然後就會有一個相對的物質和此tag有親和力可以結合。
金屬螯和層析法 (metal chelate affinity chromatography)就是一種親和層析法,在其bead上會接有一些小基團,小基團上會帶有兩個COO-group,因為COO-帶負電,這兩個負電就會去吸引帶兩個正電的nickel。我們可以在蛋白質的前面或後面加上一小段通常是6個histidine,histidine的imidazole ring上的氮 (N)帶有兩個alone pair electrons,於是nickel就可以吸附上去,蛋白質因此就可以和固相的擔體結合。最後要溶離出所要的蛋白質時就用未接有任何東西的imidazole來競爭,這是一個很溫和的方法,在溶離的過程中,蛋白質不容易變性。
因此在作實驗時,若是要表現一個蛋白質,可以把這蛋白質的基因接在6個histidine的前面或是後面,那麼表現出來的蛋白質就可以直接利用nickel colume來作純化分離。
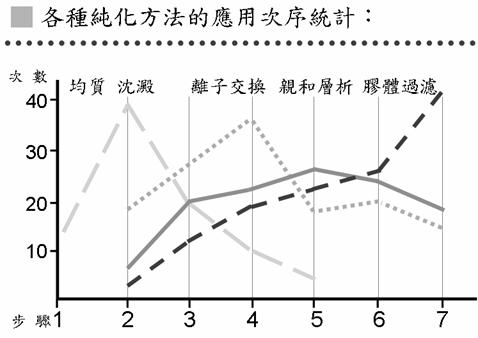 以應用次序的統計來說(圖10),第一步都是先用均質;沈澱法放在第二步使用的比較多;離子交換法則多用在第三步或第四步;親和層析法在各個步驟都有;而膠體過濾法多半放在比較後面的步驟,可能是多半利用在去除鹽纇,所以多放在後面的步驟。沈澱有時也會用在第一步,例如在做液體的樣本時,就不需要均質,可直接做沈澱。有時會同一個方法用兩次,但兩次方法的應用必須是不同目的;例如在做離子交換法時,第一次使用可以先把環境的pH值調到蛋白質的pI之上,用陰離子交換法,經過幾個步驟之後,再把pH值調到pI之下,用陽離子交換法。在實際應用上,這些純化方法都可以因目的不同作不同的組合應用。
以應用次序的統計來說(圖10),第一步都是先用均質;沈澱法放在第二步使用的比較多;離子交換法則多用在第三步或第四步;親和層析法在各個步驟都有;而膠體過濾法多半放在比較後面的步驟,可能是多半利用在去除鹽纇,所以多放在後面的步驟。沈澱有時也會用在第一步,例如在做液體的樣本時,就不需要均質,可直接做沈澱。有時會同一個方法用兩次,但兩次方法的應用必須是不同目的;例如在做離子交換法時,第一次使用可以先把環境的pH值調到蛋白質的pI之上,用陰離子交換法,經過幾個步驟之後,再把pH值調到pI之下,用陽離子交換法。在實際應用上,這些純化方法都可以因目的不同作不同的組合應用。
圖10:各種蛋白質純化方法的應用次序統計
(3)其他方法
應用在純化分離蛋白質的方法還有沉澱法(precipitation)、或者使用先進的蛋白質晶片(protein chip)技術來進行。沉澱法有免疫沉澱法(immunoprecipitation)、共沉澱(coprecipitation)等方法,免疫沉澱法是使用會和目標蛋白質以專一性結合的抗體,讓其與溶液中的目標蛋白質結合、並沉澱,來達到和溶液中其他的蛋白質分離的效果。這個方法一開始,在於如何取得專一性高的抗體,且抗體和目標蛋白質間的作用力最好是剛好能達到分離的效果、但又不會在最後純化的步驟時不易將抗體與蛋白質分開。在抗體的製備上,最直接的方法是用合成peptide來注入動物體內以生成、取得所要的抗體,又或者可設計chimeric gene product(如GST fusion protein)、tagged gene product(如His-tagged protein)等構築(construct)重組DNA (recombinant DNA)的方法來合成理想的抗原,再以此抗原注射入動物體內誘發產生抗體,此類方法唯獨有個缺點就是太過繁瑣。
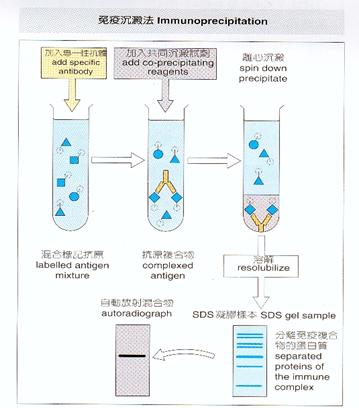 共沉澱與免疫沉澱法所使用的原理與方法大致相似,只是與目標蛋白質結合、沉澱的角色由另外一個蛋白質來取代。首先要瞭解的是蛋白質的結構是由peptide(s)
folding所組成的,其露出來的功能性區域(通常為N端)以及其它的調節區域,可以藉這些區域與其它蛋白質(如酵素、接受器、或scaffold等)結合發生反應,而找出蛋白質-蛋白質interaction的關係,讓兩者反應結合、並共同沉澱,以達初步的純化分離的目的。其大概方法為將已知會與目標蛋白質A結合作用的蛋白質B加入溶液中與蛋白質A反應,離心沉澱後再取其沉澱部分溶入緩衝液,如phosphate-buffered
saline,之後再以Western
blotting來做檢測,交替使用anti-A、以及anti-B的抗體做RIA,若是為蛋白質A-B複合體則會對此二種抗體都發生反應。然而要擔心的一個問題,就是不論在免疫沉澱法中的抗體-蛋白質結合,亦或是共沉澱中的蛋白質-蛋白質結合,在結合、反應後會不會改變其原本蛋白質的功能與構形,這是在使用沉澱法所要注意的一點。
共沉澱與免疫沉澱法所使用的原理與方法大致相似,只是與目標蛋白質結合、沉澱的角色由另外一個蛋白質來取代。首先要瞭解的是蛋白質的結構是由peptide(s)
folding所組成的,其露出來的功能性區域(通常為N端)以及其它的調節區域,可以藉這些區域與其它蛋白質(如酵素、接受器、或scaffold等)結合發生反應,而找出蛋白質-蛋白質interaction的關係,讓兩者反應結合、並共同沉澱,以達初步的純化分離的目的。其大概方法為將已知會與目標蛋白質A結合作用的蛋白質B加入溶液中與蛋白質A反應,離心沉澱後再取其沉澱部分溶入緩衝液,如phosphate-buffered
saline,之後再以Western
blotting來做檢測,交替使用anti-A、以及anti-B的抗體做RIA,若是為蛋白質A-B複合體則會對此二種抗體都發生反應。然而要擔心的一個問題,就是不論在免疫沉澱法中的抗體-蛋白質結合,亦或是共沉澱中的蛋白質-蛋白質結合,在結合、反應後會不會改變其原本蛋白質的功能與構形,這是在使用沉澱法所要注意的一點。

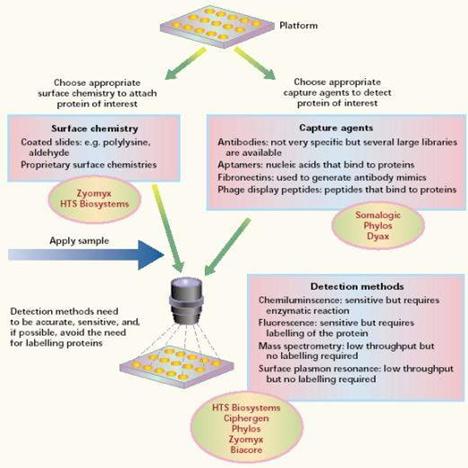
除了沉澱法外,還有利用蛋白質晶片的技術來做純化分離的工作,現今有研發出「軟微影技術」(soft lithography),是大量製作微小結構的全新方法之一,

蛋白質晶片的製備方法與基因晶片方法相似,差異處只是將作用對象改為蛋白質,亦即玻片上點陣樣品和實驗樣品皆為蛋白質(protein/peptide)。但是在實際的製作與應用卻有非常大的困難,理由如下:
(1)蛋白質的來源取得不易
(2)蛋白質容易失去活性
(3)蛋白質晶片載體製作不易
(4)如何維持晶片上蛋白質的生物活性
(5)晶片與檢體間的最佳反應條
圖13:蛋白質晶片
1.Garrett,H.,Grisham,M.,1999,Biochemistry,p514&518,2nd ed.,Harcourt college publishers.
2.http://140.112.78.220/~juang/Protein/Purification/P1.htm